
Phasing Technologies (10x Genomics Chromium and Dovetail Genomics Hi-C)
Last January, I had the privilege of attending the Plant and Animal Genome Conference XXVI in San Diego, California. My boss calls it the only essential agrigenomics conference anyone needs to attend, and understandably so, because the big names in genomics—ones I would have never thought of coming into being beyond journal article bylines—are consistent attendees of this conference. The conference was divided into workshop sessions that tackled updates and recent developments from specific fields of study. I particularly enjoyed the sessions dedicated to bioinformatics in spite of my episodic inability to grasp the highly computational discussions.
Some of the expressions that kept on cropping up all the way through the conference were “phasing” and “phased data”. Phased data refers to genotype information wherein variants occurring in the same molecule are identified; for example, genotypes that distinguish which one of the pair of chromosome (i.e. maternal or paternal genome) holds the allele/variant. Surprisingly (to me, at least), phased data is the third crucial ingredient in producing reference-quality diploid genomes aside from short and long reads [1]. This is how the three ingredients work together:
- Short-read sequencing technologies (e.g. Illumina) enable the identification of single nucleotide polymorphisms (SNPs).
- Long-range single-molecule sequencing technologies (e.g. PacBio, Oxford Nanopore) elucidate structural variants (SVs), repetitive sequences, and microsatellites.
- Phasing technologies (e.g. 10x Genomics Chromium, Dovetail Genomics Hi-C) employ sparse sequencing (vs. true long-read sequencing) for genome scaffolding and phasing, with only a modest cost increase over standard short-read sequencing.
Discussions on short-read and long-read sequencing are far too common, and thus the rest of the entry will center on the science behind phasing technologies.
10x Genomics Chromium
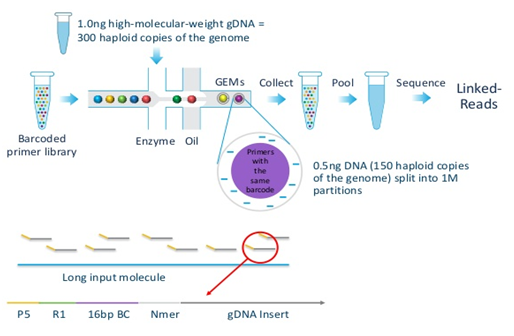
[Overview of 10x Genomics Chromium]
The technology takes advantage of a reagent consisting of several gel beads or GEM (gel bead in emulsion droplet), each of which contains several copies of a 16-base barcode unique to that bead and amplification reagents (kinda reminiscent of 454 sequencing). A microfluidic system in the form of the “Chromium Controller” partitions and delivers high-molecular-weight DNA to the beads, such that each bead contains only around 10 molecules (~0.5 Mb or 0.01% of the diploid human genome). The molecular biology of the system is arranged to produce constructs possessing the barcode, ~350 bp of gDNA from a molecule, and Illumina adapters. The constructs can be sequenced in an Illumina instrument, producing linked reads or paired reads organized by barcode and originating from the same molecule. The technology is able to generate phased data because the possibility of two gDNA molecules from the same locus with opposing haplotypes (i.e. alleles from pair of chromosomes) being present together is almost impossible [2].
Dovetail Genomics Hi-C

[Overview of Dovetail Genomics Hi-C]
Chromatin capture-based methods rely on the fact that, after fixation, DNA segments in close proximity in the nucleus are more likely to be ligated together than distant regions. Hi-C capitalizes on this method to generate sequencing libraries that include genomic regions in close spatial, but distant linear, proximity. The chromatin is crosslinked with formaldehyde, and the cross-linked chromatin is digested with a restriction enzyme that leaves a 5’ overhang. The 5’ overhang is filled with a biotinylated residue. Under a dilute condition that favors ligation events between crosslinked fragments, blunt-end fragments produce chimeric molecules. The crosslinks are reversed, and a Hi-C library is created by purifying and enriching biotin-containing fragments. A Hi-C library with proper adapters can then be sequenced in an Illumina instrument [3].
When combined with high-quality draft assemblies, data produced from these two technologies aid in the resolution of nearly entire eukaryotic chromosomes. They can also be easily integrated into existing laboratory workflows as they only need short-read sequencers in generating scaffolding and phasing data. Below are the actual papers if you want to read more about the methods:
References:
[1] Sedlazeck FJ, Lee H, Darby CA, Schatz MC. 2018. Piercing the dark matter: bioinformatics of long-range sequencing and mapping. Nature Reviews Genetics. doi:10.1038/s41576-018-0003-4
[2] Weisenfeld NI, Kumar V, Shah P, Church DM, Jaffe DB. 2017. Direct determination of diploid genome sequences. Genome Research. 27(5):757-767. doi:10.1101/gr.214874.116
[3] Lieberman-Aiden E, Van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R. 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326(5950):289-293. doi:10.1126/science.1181369
Leave a Comment